Brief Description
This study investigates the decomposition mechanism of Methyl trichlorosilane during Silicon Carbide (SiC) deposition during Chemical Vapor Infiltration (CVI). High-performance applications require SiC-based ceramic matrix composites (CMCs); however, producing them presents challenges due to limited deposition rates, high energy consumption, and uneven coating quality. To overcome these challenges, a comprehensive understanding of the SiC formation mechanism is necessary. Modeling surface reactions of MTS decomposition on the SiC substrate using density functional theory (DFT) calculations with the Vienna Ab initio Simulation Package (VASP) is the key point of current research.
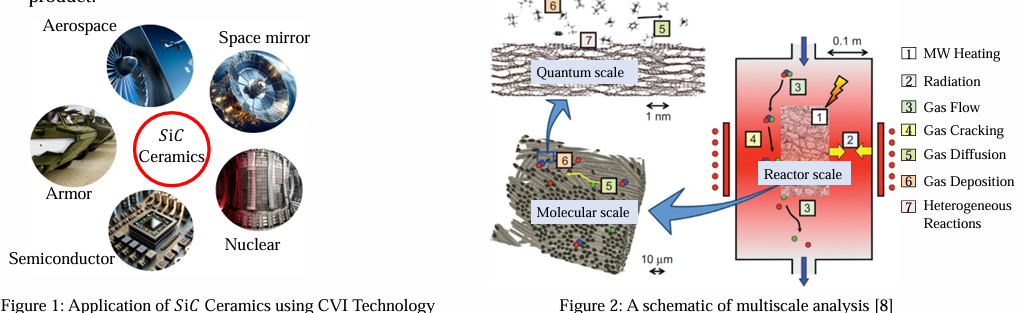

By focusing on the adsorption, reaction, and desorption mechanisms that control SiC development, our method incorporates quantum mechanical models such as Kohn-Sham equations, the Many-Body System, Born-Oppenheimer (BO) approximation, and Generalized-Gradient Approximation (GGA). Using Transition State Theory (TST) and Potential Energy Surface (PES) mapping, the study investigates reaction routes and discovers important intermediates, such as methyl (CH₃) and other hydrocarbon species. We also emphasize the function of hydrogen in surface stabilization. The findings expand our knowledge of the rate-limiting steps in MTS breakdown and offer guidance for refining CVI/CVD procedures, which could increase material quality and deposition efficiency for cutting-edge engineering applications.

Add comment
Comments
Go ahead